


By Nichollas Scott, Postdoctoral Fellow in Foster Lab
Within high-throughput screening approaches the speed of data acquisition has typically determined how much of a system researchers are able to observe. This has ensured that over the last decade the majority of analysis platforms have focused on improving depth by increasing the speed of measurements. Although this has been extremely successful in some types of analyses, the precision or discriminatory power of measurements can sometimes be more advantageous than the number of unique measurements.
In the biological sciences, precision and care are critical for the success of most experiments. Yet in high-throughput screening techniques, trade-offs in quality, type or speed are commonly required to achieve depth. Within one such high-throughput technique, mass spectrometry, these trade-offs are well known to trained mass-spectrometrists, yet rarely communicated to the broader scientific community.
Mass spectrometry (MS) is an analytical technique that allows the determination of compounds by accurate measurements of their mass. Within MS, time and quality of data are typically linked: the longer a component is measured, the higher the data quality. Importantly, when mixtures of unknown samples are analyzed, such as in proteomics or metabolomics experiments, instrumentation is typically optimized for speed. The exact speed setting is dependent on the expected components to be analyzed. Thus, rather than collecting the most informative data for all components, instruments are tuned for the fastest generation of the most information, assuming the specific components’ class. This creates a situation where the assumed components are preferentially identified, leaving unexpected components less likely to be identified due to inadequate information.
As instrumentation used for proteomics experiments are typically set up and optimized to analyze non-modified peptides derived from tryptic digestions, these peptides are far easier to identify than other peptides, which may be of biological relevance. If alternative components are being investigated, an alternative set up may be required. In fact, for some components a standard set-up will not result in the data a user desires, such as in the case of certain peptide sequences or protein modifications. In these cases, increasing the acquisition speed does not increase the likelihood of generating informative data, rather greater flexibility in the analysis of the components is required. Sometimes multiple MS approaches may be to needed generate a more complete characterization (see Case Study).
| Case study: Glycopeptides | |
|
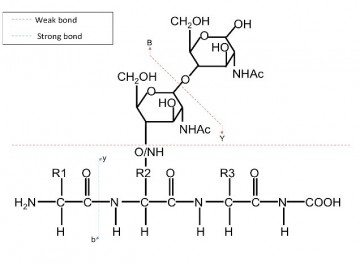
Glycopeptides are heteropolymers composed of both glycan and peptide components. The linkage strengths between the monomers of glycans and peptides differ significantly (Figure A), resulting in different fragmentation patterns depending on the fragmentation type. Glycan fragmentation dominates in approaches that utilize gentle fragmentation protocols and harsher conditions result in peptide fragmentation. By understanding this behavior, researchers can exploit this information and use multiple types of fragmentation to generate complementary information. In these situations, using two MS fragmentation approaches, one gentle and the other harsher, provides a researcher with a more complete understanding of glycopeptides. This is only one example of where speed or “depth” of the MS analysis is not required for experimental success.
|
 |
Researchers should also be aware that there is a continuous increase in the specialization of platforms in MS instrumentation. This has resulted in some vendors developing platforms that generate data extremely quickly with limited flexibility, while others opt for slower speeds with greater flexibility. It is important that users are aware of this trend, as this will determine how one may wish to analyze proteomic samples, saving time, money and sample material.
Helpful questions for researchers wishing to analyze samples using MS:
- Is the analyte similar to the samples the instrument is set up to analyze?
- If used for identification, is anything known about how the analyte of interest will fragment?
- Is quality or quantity more important? (In complex samples one may want to identify as much as possible – speed over precision but for lower complexity samples one may want to generate higher quality data for fewer identified ions – precision over speed.)