

 Proteomics aims to identify all proteins and protein forms within a biological sample such as cells or tissues. To do this, proteins are digested to peptides using a site-specific protease and analyzed within a mass spectrometer. Subsequently, matching mass spectra to protein sequence within a database identifies peptides. The specificity of the protease (i.e. the peptides sequences it cleaves) influences how well peptide sequences are determined.
Proteomics aims to identify all proteins and protein forms within a biological sample such as cells or tissues. To do this, proteins are digested to peptides using a site-specific protease and analyzed within a mass spectrometer. Subsequently, matching mass spectra to protein sequence within a database identifies peptides. The specificity of the protease (i.e. the peptides sequences it cleaves) influences how well peptide sequences are determined.
Trypsin is by far the most common protease used in proteomics and cleaves C-terminal to the amino acids Lysine and Arginine. However, not all peptides are identified using Trypsin. C-terminal peptides are rarely identified and specific phosphorylation and methylation sites are not well characterized. Thus, new digestion enzymes are needed to identify peptides carrying these modifications and also the >3800 ‘missing’ proteins which currently lack proteomics evidence of their expression in human tissues.
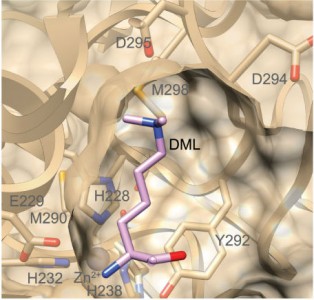
In an article recently published in Nature Methods, CBR researchers led by Dr. Christopher Overall describe the characterization of LysargiNase. A metalloproteinase from the Archaea species Methanosarcina acetivorans, LysargiNasecleaves N-terminal to Lysine and Arginine. This is the exact opposite of trypsin and so is most unusual and most useful. It offers improved identification of C-terminal peptides, methylated forms of Lysine and Arginine, and specific phosphorylation sites. Using LysargiNase in addition to Trypsin results in significantly more protein identifications. LysargiNase is an exciting new tool for the proteomic community with specific applications to studying modified protein forms.
Article contributed by Lindsay Rogers, PDF at Overall Lab, CBR